课题组动态
课题组动态
内容
光(电)催化在污染物降解方面显示出广阔的应用前景,引起高度关注。目前广泛采用的光(电)催化剂(如TiO2),主要通过界面单电荷转移及其产生的自由基物种来氧化降解污染物。这些高氧化性自由基物种往往在降解污染物的同时,也无选择性地氧化与污染物共存的有机质(如腐殖酸),严重降低了目标污染物的降解效率。另一方面,这种无选择性反应也限制了光(电)催化在有机合成方面的应用。
在国家自然科学基金委、科技部和中国科学院的大力支持下,课题组发现赤铁矿(α-Fe2O3)纳米光电催化材料表面可高效高选择性活化水分子,通过非自由基途径将水中的氧原子转移到系列有机和无机反应物上,实现了它们的高选择性氧化,从而为复杂水体中目标污染物的选择性高效去除以及化学品的高价值氧化转化提供新的策略。
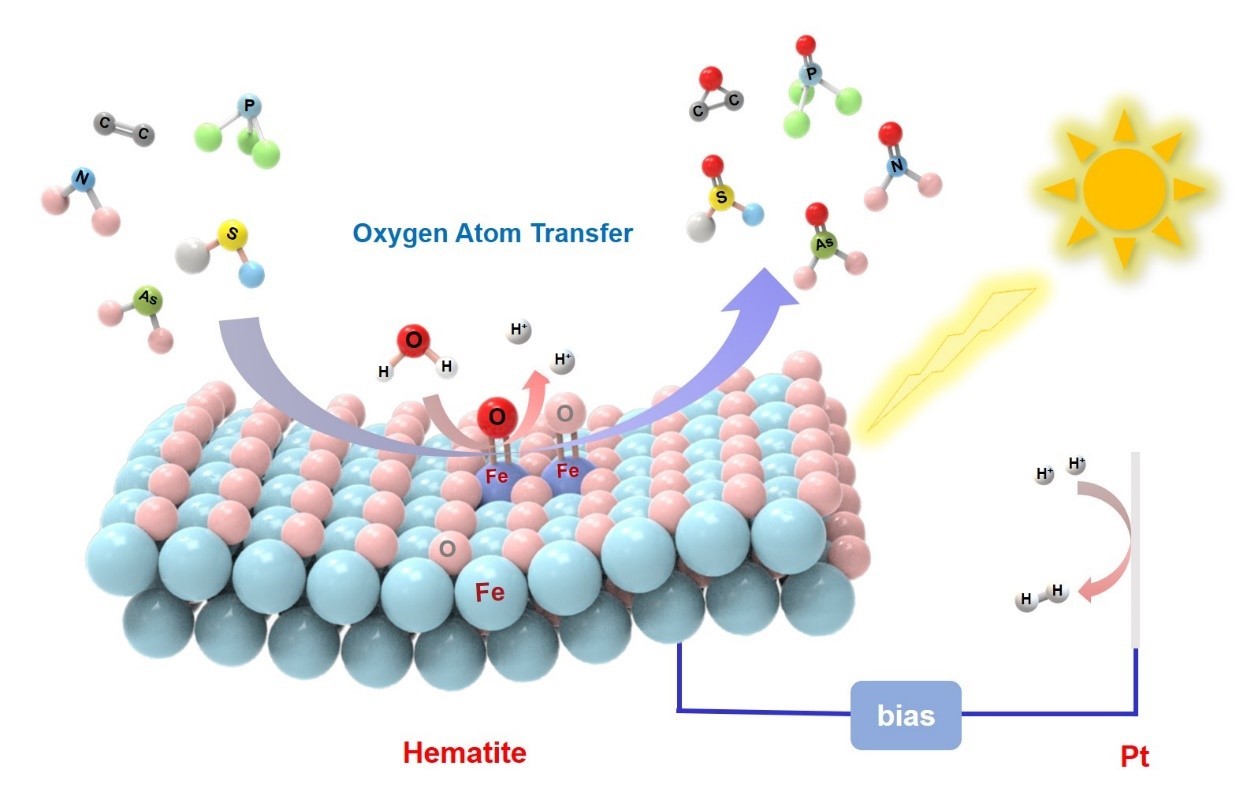
在前期研究中,我们通过动力学同位素效应、电化学阻抗谱、原位电化学红外光谱等手段,证明了α-Fe2O3表面水分子的光电催化氧化是通过水分子亲核进攻表面FeIV=O物种机制进行的,其中水分子的氧氢键断裂过程是耦合的质子/空穴转移,属于反应的决速步骤(J. Am. Chem. Soc. 2016, 138, 2705;J. Am. Chem. Soc. 2018, 140, 3264)。最近,我们进一步揭示了α-Fe2O3光电催化高选择性氧原子转移新机制,发现α-Fe2O3光电催化材料表面可高效高选择性活化水分子,通过非自由基途径将水中的氧原子转移到系列有机和无机反应物上,实现了它们的高选择性氧化,如将水中高毒性的亚硝酸盐、亚砷酸等无机污染物加氧氧化成毒性相对较低的硝酸盐和砷酸(法拉第效率分别高达86.7% 和92.0%)。对于中心原子含有孤对电子的有机物,如有机硫、有机膦等,可实现高选择性(>90%)的氧化。而在相同条件下广泛使用的TiO2光催化剂(自由基反应机理),虽然可以将苯甲酸、马来酸等有机物有效氧化,但对这些加氧反应的选择性和法拉第效率却很低。另外α-Fe2O3光电催化对这些羟基自由基敏感的有机物反应活性很低。进一步通过电化学阻抗和18O同位素示踪技术证明α-Fe2O3表面光电催化加氧过程是通过一步两空穴的氧原子转移反应进行的,而水分子是所转移氧原子的唯一来源。这一过程避免了反应选择性差的自由基路径,从而实现了高选择性氧化加氧反应。DFT计算表明α-Fe2O3和TiO2光电催化机制的不同源于二者迥异的表面电子结构。在α-Fe2O3表面的光生空穴主要分布在由Fe 3d和O 2p轨道组成的能级上,形成高价铁氧物种(FeIV=O),这些物种倾向于和氧原子受体发生氧原子转移反应;而TiO2的表面空穴处于O 2p轨道上形成Ti-O·物种,更易于发生单电荷转移,而引发自由基反应。本研究突破了传统自由基型光(电)催化反应的瓶颈,为高选择性光(电)催化提供了一种新思路(Nature Catalysis, 2021, 4, 684-691)。